

| Dr. Pilar Giraldo |
El 5 noviembre la FDA estadounidense concedió la Revisión Prioritaria de NDA (Nueva Aplicación de Medicamento) para la velaglucerasa alfa, principio activo de la terapia de sustitución enzática para el tratamiento de la Enfermedad de Gaucher tipo 1. Esto significa que se acelera el calendario de fechas de examen del medicamento de diez a seis meses. No estamos acostumbrados a estos ‘saltos’ burocráticos pero el laboratorio donde se fabrica el tratamiento más utilizado para elinar el glucocerebrósido acumulado en hígado, bazo y hueso tuvo que ‘interrumpir temalmente‘ la producción. ‘Se prevé que la escasez se resuelva a finales de 2009‘ pero, hasta entonces, existe riesgo de que se produzcan retrasos en la distribución de los pedidos. La Taliglucerasa es otra enza que estaba en ensayo clínico y también ha acelerado su procediento para darle prioridad al mecanismo y que los Pacientes puedan disponer alternativas lo antes posible.
AGENCIA ESPAÑOLA DEL MEDICAMENTO
La Enfermedad de Gaucher es una enfermedad genética, de baja prevalencia (1/100.000 habitantes), multisistémica, grave y de complicado diagnóstico. Está causada un defecto en el gen de la enza bglucocerebrosidasa, que altera las funciones normales del hígado, bazo, huesos y médula ósea. También se pueden ver afectados el riñón, el corazón, los ojos o los pulmones. No está ligada al sexo y puede manifestarse a cualquier edad; algunos grupos éticos tienen mayor prevalencia. Estas personas no disponen de cantidades suficientes de bglucocerebrosidasa y las células se llenan de grasa no digerida.
 Quizá el médico francés Philippe Charles Est Gaucher no fue consciente en 1882 de que su hallazgo cambiaría la vida de miles personas a partir de eso momento. Tras evaluar el cadáver de una mujer de 32 años de edad descubrió que su bazo era demasiado grande. A partir de entonces estos Pacientes tienen un ‘DNI patológico’ y los médicos saben a qué enfrentarse cuando diagnostican los preros síntomas.
Quizá el médico francés Philippe Charles Est Gaucher no fue consciente en 1882 de que su hallazgo cambiaría la vida de miles personas a partir de eso momento. Tras evaluar el cadáver de una mujer de 32 años de edad descubrió que su bazo era demasiado grande. A partir de entonces estos Pacientes tienen un ‘DNI patológico’ y los médicos saben a qué enfrentarse cuando diagnostican los preros síntomas.
ERNEST GAUCHER (1854 1918), historia de la medicina
Lidia Són tenía nueve años cuando fue diagnosticada de una enfermedad genética que sus padres no sufren, ni ahora sus hijos tampoco. Desde los seis años llamó a muchas puertas que no tuvieron respuesta para ella, hasta que un día fue diagnosticada de una de las más de 5.000 enfermedades raras que existen en el mundo. La suya es la Enfermedad de Gaucher, pero su historia no dista demasiado de la que cuentan los más tres millones de españoles afectados otras patologías poco prevalentes. ‘Mis padres me llevaron de médico en médico, visité cientos de sitios antes de ser diagnosticada que ellos no sufren la enfermedad y hemos acudido a Unidades de Consejo Genético para que mis hijos tampoco la sufrieran‘.
En la mayoría de los casos desde que aparecen los preros síntomas hasta el diagnóstico pasan cinco años. Para Lidia pasaron ¿sólo? tres, ¿una chica con suerte? El 20% de los Pacientes tardan más de 10 años en ser diagnosticados. ‘El día que me diagnosticaron la enfermedad fue uno de los mejores de mi vida que ya sabía contra qué tenía que luchar’. Para Lidia ‘vivir con esta enfermedad ahora es más llevadero pero fue muy difícil cuando mis hijos eran pequeños. Con el prero sólo pude jugar a hacer puzles que no tenía fuerzas para moverme y el segundo lo he disfrutado más’.
Vive en Gerona pero viaja a Zaragoza dos veces al mes para encontrarse con su médico, la Hematóloga del Hospital Miguel Servet, la Doctora Pilar Giraldo. En nuestro país la mitad de los afectados alguna de las enfermedades denominadas raras ha tenido que viajar fuera de su provincia en los dos últos años, y cerca del 40% lo ha hecho más de cinco veces. El 17%, aunque lo necesitaba, no ha podido.
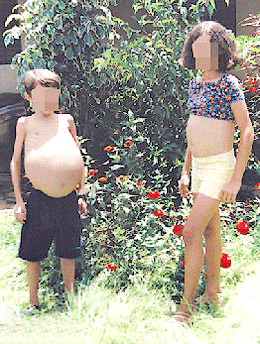 |
|
(foto ilustrativa. no tiene relación con el testonio) |
‘Era como una niña pequeña y embarazada, no tenía fuerzas para nada‘. El médico de cabecera de Lidia se dio cuenta de que el bazo le pesaba cuatro kilogramos y el prer paso fue una operación para extirpárselo. Pertenece al grupo de Gaucher tipo 1, el más común. Afecta a uno de cada 40.000 a 60.000 niños nacidos vivos y se caracteriza el aumento de tamaño del hígado y el bazo. La esperanza de vida para estas personas es de 60 años aproxadamente y son el 80% de los Pacientes de Gaucher en España.
Algunos Pacientes tipo 1 carecen de síntomas, pero para otros son severos y pueden resultar fatales. ‘Cuando la enfermedad no afecta a otros órganos es más fácil de llevar, pero a medida que te haces mayor se complica‘, asegura Són.
Explica a prsalud con lágras en los ojos las lecciones que le ha dado la vida: ‘he aprendido a valorar cada instante, para mí la felicidad es no tener dolor‘. Diagnosticarla fue el principio de todo. ‘Cuando me diagnosticaron y me trataron empecé a estar mejor, sobre todo desde que me cambiaron a Velaglucerasa‘. Un 42% de los Pacientes con alguna enfermedad rara no recibe ni apoyo ni tratamiento y un 27% recibe un tratamiento inadecuado. Menos de la mitad afirma disponer del tratamiento que precisa.
‘No somos raros, somos enfermos luchadores‘, reivindica . ‘Conozco mis límites y descanso cuando no puedo más y a la gente splemente le digo que tengo un catarro o que estoy indispuesta‘. A Lidia no le gusta dar explicaciones que le gustaría la gente conociera sus enfermedad, ‘pienso mucho en los padres desesperados que van puerta tras puerta haciendo preguntas, igual que los míos hicieron en su día’. Ahora ‘aguanto mucho más el esfuerzo físico, el trabajo, el día a día. La gente me dice que tengo un aspecto radiante‘. Al menos ella tiene un final feliz.
Seguiremos Informando…









